Primary Plasma Cell Leukaemia (pPCL) is a very rare and aggressive subtype of plasma cell dyscrasias with a very poor outcome and characterized by presence of >2x109/l circulating plasma cells. Overall incidence being 1%-2% of all malignant plasma cell diseases and 0.9% of all acute leukaemias. Patients with primary and secondary PCL have similar clinical features but differences do exist. Here we report a rare and interesting case diagnosed as primary PCL in a 44-year-old female with unusual clinical presentation and complex karyotyping. Her serum electrophoresis showed a monoclonal IgG component and immunohistochemistry of bone marrow plasma cells showed CD 38 positive and CD 20 negative. The patient initially responded partially for combination chemotherapy, but succumbed after 20 days of diagnosis. We are presenting this case to highlight the importance of early diagnosis of such haematological malignancies in settings where treatment options like stem cell transplantation are limited.
Acute renal failure, Cytogenetics, Prognosis
Case Report
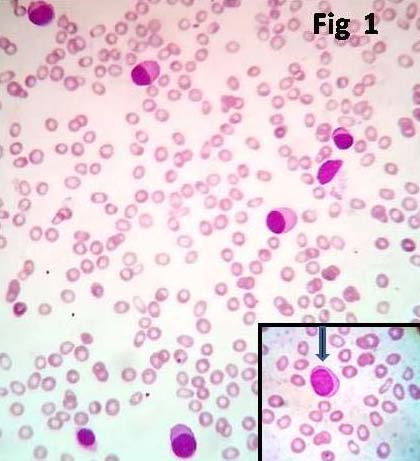
A 44-year-old female with history of Type II diabetes mellitus and severe anaemia presented to emergency department with vomiting, nausea and abdominal pain. General examination revealed severe pallor and systemic examination revealed hepatosplenomegaly. There was no lymphadenopathy. Laboratory data showed haemoglobin 6.5 gm%, WBC count was 20x103/µl, peripheral smear examination showed rouleaux formation with microcytes hypochromia and anisopoikilocytosis with 40% plasma cells and 27% plasmablasts [Table/Fig-1]. Urine examination showed 4+ protein, 2+ leukocyte esterase and positive Bence Jones protein test.
Microphotograph of peripheral smear showing many plasma cells having eccentrically placed nuclei and abundant basophilic cytoplasm (x100, Leishman stain). Inset: arrow showing plasmablast (x1000, Leishman stain).
Liver and renal function tests showed hypoalbuminaemia, hyperuricaemia and hyperphosphataemia, blood urea 106 mg/dl, serum creatinine 4.8 mg/dl, serum calcium 12 mg/dl, Lactate Dehydrogenase (LDH) 450U/l, ESR 36 mm/hr, and B2 microglobulin>20,000 ng/ml.
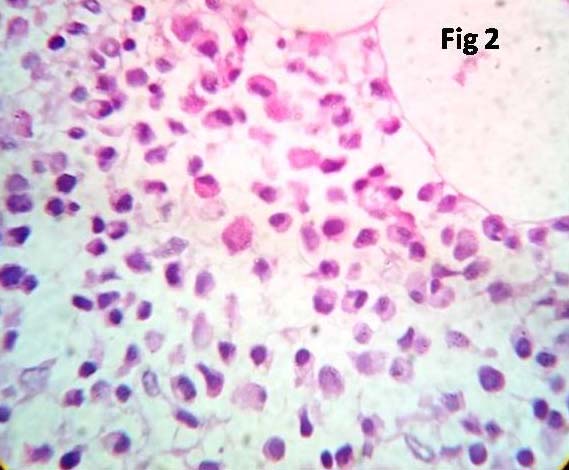
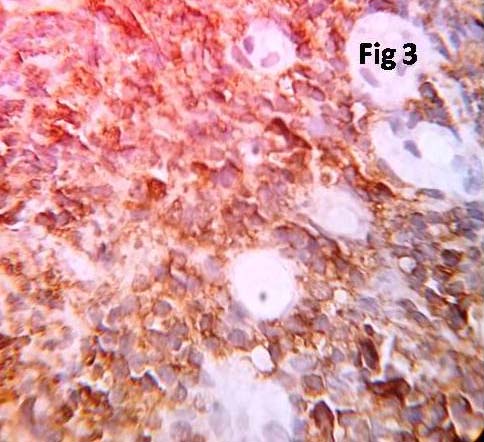
Serum protein electrophoresis showed a monoclonal band in the gamma region. X-ray of skull and spine showed only osteoporosis and did not show any osteolytic lesions. Bone marrow aspiration and biopsy were performed. Aspirate revealed diluted sinusoidal blood. Bone marrow biopsy showed mostly cortical bone with small groups of plasma cells amidst few erythroid series cells [Table/Fig-2]. Immunohistochemistry of marrow biopsy reveled plasma cells expressing CD 38 positivity [Table/Fig-3] and CD 20 negativity.
Microphotograph of bone marrow biopsy showing sheets of plasma cells replacing the normal marrow cells (x 400, H&E).
Immunohistochemical staining for CD38 highlighting plasma cells (x400).
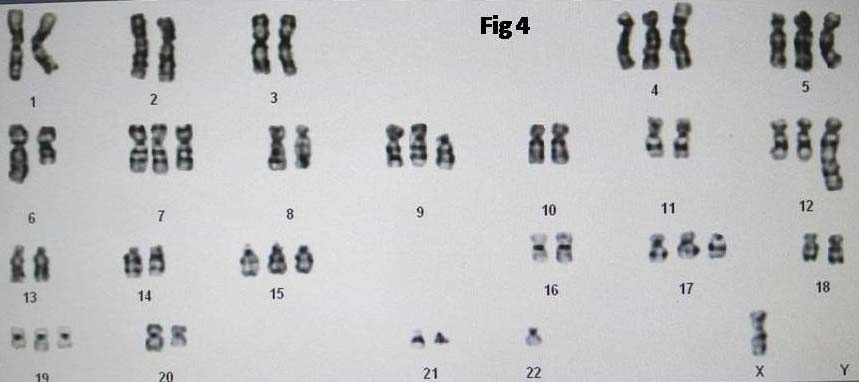
Karyotyping was done on blood sample which showed chromosomal abnormality i.e., hyperploidy with monosomy X(51,XO). The following abnormalities were noted:
51,X,-Xadd(4)(p16),+5,del(6)(q21),+7,+8,add(8)(q24)x2,+del(9)(p11),+12,dict(1;12)(p11;q24),+19,-22,+mar [Table/Fig-4].
Chromosomal analysis of peripheral blood showing a complex karyotype.
Our patient was diagnosed to have primary PCL based on the findings of peripheral smear, bone marrow biopsy, serum electrophoresis and cytogenetic analysis. After first cycle of chemotherapy, plasma cell count reduced to 25% and plasmablasts to 12%. Later, her conditions deteriorated and succumbed within 20 days of diagnosis due to uraemic encephalopathy. Consent was not given for clinical autopsy by patient’s relatives.
Discussion
PCL is defined as a clonal proliferation of plasma cells first diagnosed in the leukaemic phase [1]. The first cases of PCL were first described in a 63-year-old woman at autopsy, who had widespread infiltration of plasma cells in multiple organs and a 47-year-old man with Multiple Myeloma (MM) developed terminal leukaemia with atypical plasma cells [2].
The median age group for primary PCL is usually above 50 years, but few case reports in the literature are present where PCL is seen in very young age [3,4]. The male to female sex ratio distribution in both primary and secondary PCL is 3:2 [5]. The main clinical symptoms of primary PCL are asthenia, severe anaemia, thrombocytopenia, hepatomegaly, spleenomegaly, renal impairment, lymphadenopathy, pleural effusion etc., whereas higher incidence of bone lesions and lower incidence of extramedullary symptoms are seen in secondary PCL. [6] Renal involvement in haematologic disorder like MM is known [2,7,8] but very few cases of PCL with renal involvement have been reported in the previous literature. Among the various clinical presentation, acute renal failure and heavy proteinuria may be the presenting symptoms as seen in our case.
Cytogenetic abnormalities are a common feature of PCL, with 70% seen in primary PCL and 100% in secondary PCL. In primary PCL, very high incidence of chromosome 13 monosomy is seen which is associated with a short survival. The various structural abnormalities include chromosome 14q32 translocation with various rearrangements like t(6;14)(p21.1;q32.3), t(11;14)(q13;q32.3), t(14:18)(q32.3;q21.3) and many more. The 14q32 translocation is associated with leukaemic transformation in MM [6]. In one of the series, out of 26 cases which were published by Gracia-sanz et al., 22 cases showed diploid or hypoploid karyotype [9]. In another study done by Avet-Loiseau H et al., 23/24 patients showed pseudodiploid or hypodiploid karyotype which is seen in 50% cases of MM [10].
Other abnormalities detected are monosomy 7, trisomy 18, and monosomy X in women. Monosomy X was seen in our case too which is again a marker for poor prognosis. Case series done by Gracia-Sanz R et al., showed monosomy X in 25% of their PCL cases [9].
The various prognostic parameters of PCL include a low serum albumin, hypercalcaemia, elevated beta 2 microglobulin, serum C-reactive protein, serum LDH, absolute peripheral plasma cell count of >4x109, thrombocytopenia, increased percentage of S-phase of plasma cells, advanced age (>60 years/older), poor performance status (Eastern cooperation oncology group grade 2) etc., [3].
The various differential diagnoses for presence of plasma cells in peripheral smear include reactive processes which may be secondary to bacterial or viral infections like parvovirus B19, hepatitis, Epstein-Barr virus, dengue or autoimmune conditions like rheumatoid arthritis, systemic lupus erythematosus, Sjögren’s syndrome etc. Hence, bone marrow aspiration or biopsy helps in establishing the diagnosis of PCL [8].
PCL needs to be treated aggressively as acute leukaemias [6]. Prognosis and survival are known to be poor in PCL, with 28% of patients dying in the first month following diagnosis. Therapy initiation should begin promptly and aim for rapid disease control which will prevent early death. Chemotherapy agents like bortezomib based regimens are recommended followed by autologous stem cell transplantation, if feasible [3]. Few case reports of PCL with longest survival after treating with high dose of combination of chemotherapy drugs along with autologous stem cell transplantation have also been documented [2,11,12]. This suggests the importance of stem cell transplantation in all cases of PCL. Our patient received one cycle of induction therapy with bortezomib, thalidomide and dexamethasone as autologous stem cell transplantation was not possible.
Conclusion
This case was presented because primary PCL is a rare entity with an aggressive behaviour which has very poor outcome. Complex karyotype is an indicator of poor prognosis in primary PCL. Though renal involvement is seen in MM, one may miss the diagnosis of PCL if it has unusual presentation such as acute renal failure as seen in our case. This highlights the need for an early recognition of plasma cells in the peripheral smear and initiate for treatment with chemotherapy agents in settings where treatment options like stem cell transplantation are limited and delayed diagnosis will have a negative impact on the prognosis.
[1]. Leite LAC, Magalhães AG, Silva JF, Silva AP, Medeiros PL, Acute abdominal pain revealing a primary plasma cell leukaemia: a rare and aggressive case of plasma cell dyscrasiaJ Cytol Histol 2015 6:364 [Google Scholar]
[2]. Saccaro S, Fonseca R, Veillon DM, Cotelingam J, Nordberg ML, Bredeson C, Primary plasma cell leukaemia: Report of 17 new cases treated with autologous or allogenic stem cell transplantation and review of the literatureAm J Haematol 2005 78(4):288-94. [Google Scholar]
[3]. Marshall R, Vaughan J, David R, Schapkaitz E, Carmona S, Wiggill T, Primary plasma cell leukaemia in a 22-year-old woman: A case reportAfr J Lab Med 2015 4(1):5 [Google Scholar]
[4]. Raj RS, Najeeb S, Aruna R, Pavithran K, Thomas M, Primary plasma cell leukaemia occurring in the youngIndian J Cancer 2003 40:116-17. [Google Scholar]
[5]. Alghasham N, Alnounou R, Alzahrani H, Alsharif F, Plasma cell leukaemia: Clinicopathologic, immunophenotypic and cytogenetic characteristics of 4 casesHaematol Oncol Stem Cell Ther 2015 8(2):71-77. [Google Scholar]
[6]. Goyal M, Mohammad N, Palanki SD, Vaniawala SN, Primary plasma cell leukaemia with light chain secretion and multiple chromosomal abnormalities: How successfully treated?–A case report with review of literatureIndian J Med Paediatr Oncol 2010 31(3):96-100. [Google Scholar]
[7]. Podduturi V, Mohmand H, Gill J, Zhou XJ, Plasma cell leukaemia presenting as acute kidney injury with heavy proteinuriaWorld J Nephrol Urol 2014 3(3):129-33. [Google Scholar]
[8]. Bansode YV, Admane V, Goroshi M, Katria P, Plasma cell leukaemia a rare cause of disproportionate anemia in a patient presenting as CKDJ Assoc India 2014 62:271-73. [Google Scholar]
[9]. Garcia-Sanz R, Orfao A, Gonzalez M, Tabernero MD, Blade J, Moro MJ, Primary plasma cell leukaemia: clinical, immunophenotypic, DNA ploidy and cytogenetic characteristicsBlood 1999 93(3):1032-37. [Google Scholar]
[10]. Avet-Loiseau H, Daviet A, Brigaudeau C, Callet-Bauchu E, Terrӑ C, Lafage-Pochitaloff M, Cytogenetic, interphase and multicolour fluorescence in situ hybridization analysis in primary plasma cell leukaemia: a study of 40 patients at diagnosis, on behalf of the Intergroupe Francophone du Myӑlome and the Groupe Francais de Cytogenetique HematologiqueBlood 2001 97:822-25. [Google Scholar]
[11]. Sekiguchi Y, Shimada A, Wakabayashi M, Sugimoto K, Tomita S, Izumi H, A case of secondary plasma cell leukaemia resistant to novel agents, in which stringent complete remission was achieved and maintained for a long period of time after VAD therapy and tandem autologous transplantationInt J Clin Exp Pathol 2014 7(9):6313-22. [Google Scholar]
[12]. Wiernik PH, Sciortino D, Paietta E, Papenhausen P, Ciobanu N, Roberts M, Plasma cell leukaemia with an unususal karyotype and prolonged survival with oral alkylating agent therapyJ Cancer Res Clin Oncol 1987 113(6):576-78. [Google Scholar]