Prenatal Screening for Rare Co-Inheritance of HbE and β-Thalassaemia Traits in Western India
Parth S Shah1, Hari Shankar P Ray2, Ketan K Vaghasia3, Sandip C Shah4, Mandava V Rao5
1 Chief Scientific Officer (CSO), Molecular Genomics, Supratech Micropath Laboratory and Research Institute, Ahmedabad, Gujarat, India.
2 Research Scientist, Supratech Micropath Laboratory and Research Institute, Ahmedabad, Gujarat, India.
3 Senior Scientist, Supratech Micropath Laboratory and Research Institute, Ahmedabad, Gujarat, India.
4 Laboratory Director, Supratech Micropath Laboratory and Research Institute, Ahmedabad, Gujarat, India.
5 Ex. Director, School of Sciences, Gujarat University, Ahmedabad, Gujarat, India.
NAME, ADDRESS, E-MAIL ID OF THE CORRESPONDING AUTHOR: Dr. Sandhip C Shah, Supratech Micropath Laboratory and Research Institute, Kedar Complex, Opp. Krupa Petrol Pump, Near Parimal Garden, Ahmedabad-380006, Gujarat, India.
E-mail: supratech18@gmail.com
The mutations in Haemoglobin Beta (HBB) gene, bring about less or no production of Hb β-chain synthesis in affected cases, leading from minor to major types depending on haematological indices. In compound heterozygotic conditions, two traits are involved, in which one parent has HbE trait and the other has β-thalassaemia carrier (trait). Here, we report a family of Rajasthan, West India which had a proband (son) having HbE/ β-thalassaemia a co-inherited compound heterozygosity as revealed by DNA sequencing. It also contained upper levels of HbE with altered Hb and red cell indices showing asymptomatic to symptomatic state requiring blood transfusion periodically. The parents and Chorionic Villus Sampling (CVS) were HbE and β-thalassaemia traits only. Such case is rare in Western India and we recommend this family for genetic counseling and genetic testing before they want reproductive choices in future for better management in a society.
Compound heterozygosity, Electrophoresis, Genetic testing, Phenotypic indices, Sequence analysis
Case Report
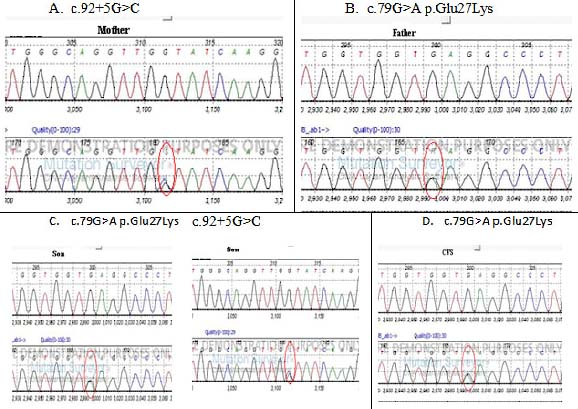
A family in Rajasthan attended a clinic as the proband (son) confessed β-thalassaemia symptoms because of low Hb levels, altered Mean Corpuscular Volume (MCV) and Mean Corpuscular Haemoglobin (MCH) values as reported by clinician. Then this Rajasthani family was referred for genetic testing to our Research Laboratory Ahmedabad. They were asked to fill consent forms. They have two probands, i.e., son and CVS. The blood of trio samples and CVS were subjected to electrophoresis for Hb variants and other blood indices. In this study CVS was suggested by clinician as it is better technique detected earlier than amniocentesis, which helps to identify the abnormal genetic condition after 8-10 weeks of pregnancy. Eukaryotic DNA was extracted and was subjected to Sanger’s DNA sequencer with appropriate primers. We detected that father had HbE trait (c.79 G>A) and mother was HBB: c.92 +5 G>C (β0) trait. However sequence analysis of proband revealed the son had combined c.79 G>A (HbE) and C 92 +5 G>C (β+) heterozygotic state, whereas CVS exhibited only c.79 G>A (HbE trait) mutation only. Phenotypic parameters indicated mild to moderate anemic conditions as suggested by a clinician, where HbE levels were high (91.3%) along with an altered red cell indices like MCV and MCH values followed by low Hb levels [Table/Fig-1 and 2].
Haematological indices of family (Rajasthani), MCV= Mean corpuscular volume, MCH = mean corpuscular haemoglobin, Hb= Haemoglobin, CVS= chorionic villus sampling.
| Sample | Haemoglobin indices | Units | Normal Range | Mutations | Genotype | Inference/Clinical Report |
|---|
| Mother (35) | Hb | 9.4g/dL | 12 to 15 g/dL | c.92+5G>C | β+ / β | Thalas-saemia Minor |
| HbA2 | 5.9% | 1.5 to 3.5 % |
| MCV | 72.1fL | 80 to 96 fL |
| MCH | 19.2pg | 33 to 36 pg |
| Father (37) | Hb | 10.5g/dL | 13 to 17 g/dL | c.79G>A p.Glu27Lys | HbE / β | HbE Trait |
| MCV | 70.8fL | 80 to 96 fL |
| MCH | 20.3pg | 33 to 36 pg |
| HbE | 71.40% | Absent |
| Proband/Son (5) | Hb | 6.25g/dL | 13 to 17 g/dL | c.79G>A p.Glu27Lys and 92+5G>C | HbE/β+ | HbE/ β – Thalass-aemia* |
| MCV | 62.2fL | 80 to 96 fL |
| MCH | 20.1pg | 33 to 36 pg |
| HbE | 91.3% | Absent |
| CVS | | - | - | c.79G>A p.Glu27Lys (HbE) | HbE / β | HbE Trait |
Figures in Parenthesis indicate age in years. * Clinician suggested periodic transfusion.
Showing DNA sequence analysis of family (Rajasthani); A. Mother with c.92+5G>C mutation (β+). B. Father with c.79 G>A P.Glu 27 lys (HbE trait), C. (Son) had (HbE, β+) co-inherited with/β-thalassaemia. D. CVS with HbE trait only. Nucleotide color, A= Green, T = Red, G= Black, C= Blue.
Discussion
Thalassaemias of α and β types are blood borne genetic anomalies. These are autosomal recessive disorders inherited from one generation to other affecting children. The β-thalassaemia is dominant in Middle East, Mediterranean region, Asia, India, Africa and Far East [1-4]. About 3-17% of population is affected by β-Thalassaemia in Indian sub continent [5].
The β-thalassaemia minor (trait) co-exists with several other traits like HbD, HbE, HbS etc to become combined heterozygotic condition having minor, intermedia and major thalassaemia depending on phenotypic indices. The HbE/β-thalassaemia condition was more in North-Eastern Indian population and 50% of them have β-thalassaemia major. These patients presented a variable clinical futures presenting non-severe to severe anemic conditions requiring blood transfusions [6]. Agarwal S et al., detected high frequency of HbE/β-thalassaemia mutation (57%) in (21) families of North India [7]. Mondal and Mandal detected only 1.16% double heterozygous state in West Bengal [8]. But in Western India this condition is rare where, in a Gujarati family, co-inheritance of HbE/β-thalassaemia was reported with sickle cell anemia disease only [9]. In an exhaustive study documented by Vichinsky EP et al., HbE/β-thalassaemia predominated in Western America [2]. These variations could be due to several factors like interplay between qualitative and quantitative levels of Hb, Iron load, geographic distribution and gene polymorphism [7,10]. However, in our study also a rare condition of HbE/ β double heterozygous condition was detected in a Rajasthani family of Western India. The proband son was affected with such type of heterozygosity but the other CVS.
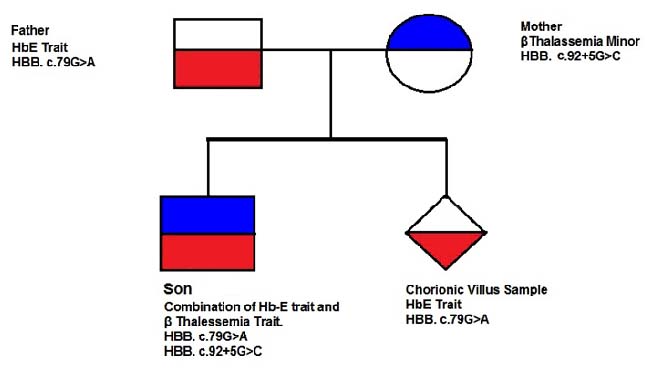
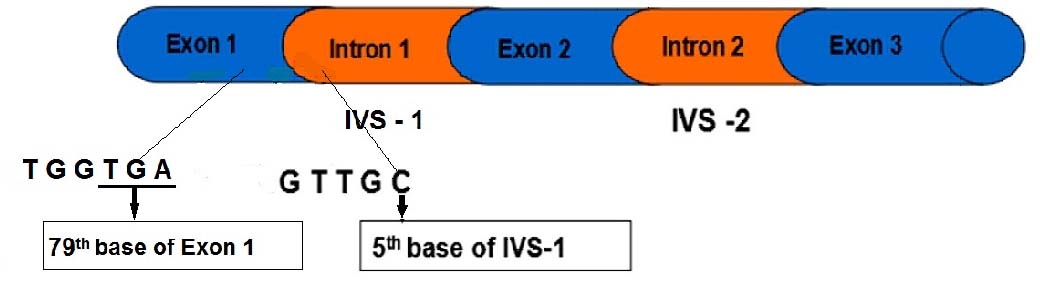
Sample exhibited only HbE trait. The son had this transmission, i.e., HBB c.79 G>A (HbE) and HBB c.92 +5 G>C (β+) from father and mother respectively and may require periodic blood transfusion as it had high HbE levels and altered normal Hb level (6.25 g/dl) along with red cell markers like MCV and MCH percent values as mentioned above. Clinician also suggested thalassaemia intermedia condition for periodic blood transfusion due to its probable severs condition. This kind of E/β compound heterozygosity is rare in Western India except one case in Gujarat [9] as for our knowledge is concerned in literature. But our case is the first to report from Rajasthan state reflecting its low incidence in this region too. Further, it is recommended that HbE/β thalassaemia patients do exhibit mild to severe anemic conditions variable from patient to patient availing periodic transfusions from childhood. It follows pedigree analysis showing autosomal recessive inheritance pattern [Table/Fig-3]. Molecular analysis indicated that both mutations of HbE and β thalassaemia in proband (son) are presented in Exon1 and Intron 1 of HBB gene whose segregation is slow probably for its low frequency [Table/Fig-4].
Pedigree chart depicting autosomal recessive inheritance pattern of gene mutation transmission in a family.
Showing Structure of HBB gene with mutation location.
Conclusion
Co-inheritance of HbE-β thalassaemia case reported in a family of Rajasthan is rare in Western India. The proband (son five years) requires periodic blood transfusions as HbE level was higher with low Hb levels and changed phenotypic indices leading to thalassaemia intermedia condition. But parents and CVS possessed HbE and β thalassaemia traits only. Such families are suggested for prenatal screening for betterment in the society and to discourage marriages between such carriers.
Figures in Parenthesis indicate age in years. * Clinician suggested periodic transfusion.
[1]. Modell B, Darlison M, Global epidemiology of haemoglobin disorders and derived service indicatorsBull World Health Orgn 2008 86(6):480-87. [Google Scholar]
[2]. Vichinsky EP, MacKlin EA, Waye JS, Lorey F, Olivieri NF, Changes in the epidemiology of thalassaemia in North America: A new minority diseasePediatrics 2005 116(6):818-25. [Google Scholar]
[3]. Fucharoen S, Weatherall DJ, The haemoglobin E thalassaemiasCold Spring Harb Perspect Med 2012 :a011734 [Google Scholar]
[4]. Weatherall DJ, Clegg JB, Thalassaemia Syndromes 2001 4th edOxfordBlackwell Scientific Publications:1-846. [Google Scholar]
[5]. Patel AP, Naik MR, Shah MM, Sharma NP, Parmar PH, Prevalence of common haemoglobinopathies in Gujarat: An analysis of a large population screening programNational Journal of Community Medicine 2012 3(1):112-16. [Google Scholar]
[6]. Mukherjee MB, Nadkarni AH, Gorakshakar AC, Ghosh K, Mohanty D, Colah RB, Clinical, haematologic and molecular variability of sickle cell β-thalassaemia in western IndiaInd J Human Gene 2010 16(3):154-58. [Google Scholar]
[7]. Agarwal S, Gulati R, Singh K, Haemoglobin E-Beta thalassaemia in Uttar PradeshIndian Pediatri 1997 34:287-92. [Google Scholar]
[8]. Mondal SK, Mandal S, Prevalence of thalassaemia and haemoglobinopathy in eastern India: A 10-year high-performance liquid chromatography study of 119,336 casesAsian J Transfus Sci 2016 10(1):105-10. [Google Scholar]
[9]. Dhawan D, Chaudhury S, Chandratre K, Ghosh A, Sojitra N, Hirapara S, Prenatal screening for co-inheritance of Sickle cell Anemia and β -Thalassaemia traitsChlin Med Biochem 2016 2(1):10000108 [Google Scholar]
[10]. Olivieri NF, Pakbaz Z, Vichinsky E, Hb E/beta-thalassaemia: a common & clinically diverse disorderIndian J Med Res 2011 134(4):522-31. [Google Scholar]