Clinical and Genetic Analysis of Fibrodysplasia Ossificans Progressiva: A Case Report and Literature Review
Maheshwar Lakkireddy1, Vijaykrishna Chilakamarri2, Prajnya Ranganath3, Abhishek Jagdishchander Arora4, Maria Celestina Vanaja5
1 Assistant Professor, Department of Orthopaedics, Nizam’s Institute of Medical Sciences, Hyderabad, India.
2 Professor, Department of Orthopaedics, Nizam’s Institute of Medical Sciences, Hyderabad, India.
3 Assistant Professor, Department of Medical Genetics, Nizam’s Institute of Medical Sciences, Hyderabad, India.
4 Assistant Professor, Department of Radiology, Nizam’s Institute of Medical Sciences, Hyderabad, India.
5 Research Associate, Diagnostics Division, Centre for DNA Fingerprinting and Diagnostics, Hyderabad, India.
NAME, ADDRESS, E-MAIL ID OF THE CORRESPONDING AUTHOR: Dr. Maheshwar Lakkireddy, Flat No: A03-505, Sadbhavana Township, APRSCL, Rajiv Swagruha, Pocharam (Village), Ghatkesar (Mandal), Rangareddy (District), Telangana-500088, India.
E-mail: maheshwar.ortho@gmail.com
Fibrodysplasia ossificans progressiva (FOP) is a rare genetic disorder characterized by congenital malformation of the great toes and disabling heterotopic ossification in specific anatomic locations with a world wide prevalence of 1 in 2 million population. Nearly 90% of patients with FOP are misdiagnosed and mismanaged. We present a case of a four-year-old boy brought by his parents with the complaints of stiffness of right shoulder, neck and multiple swellings over the upper back noted over the past 4 months. On examination bilateral symmetrical hallux valgus with microdactyly of great toes and multiple bony hard swellings on both the scapulae were noted. Skeletal survey revealed all the classical features of FOP. Mutation of the ACVR1gene on genetic analysis confirmed the diagnosis of FOP. Invasive surgical procedures including biopsy and manipulations for stiff joints were avoided as they strikingly end up in rapid progression of FOP. Congenital hallux valgus with short great toe in a child should be considered as an early diagnostic tool for FOP even before the onset of mass lesions. Genetic analysis for mutation of ACVR1gene is confirmatory. Prevention of injury, medical management of acute painful flare-ups and rehabilitation are the mainstay of treatment.
ACVR1 gene, Congenital hallux valgus, Heterotopic ossification, Iatrogenic Injury, Myositis ossificans progressiva
Case Report
A four-year-old right hand dominant boy was brought with the complaints of stiffness of right shoulder and neck, multiple swellings over the upper back noted over the past 4 months. The symptoms were insidious in onset and rapidly progressive. There was no history of preceding trauma or illness. He had been examined by various doctors previously and was extensively evaluated with no conclusive diagnosis. As the symptoms persisted the child was referred to our hospital for evaluation and management.
The child was the first born offspring of a non-consanguinous couple with no history of similar illness in any family member. On examination he had bilateral symmetrical hallux valgus with microdactyly of the great toe [Table/Fig-1]. Genu valgum was noted in both the lower limbs. Bony hard swellings were noted in anteromedial aspect of both the proximal tibiae [Table/Fig-2]. Multiple bony hard swellings of variable sizes were noted on both the scapulae. A bony hard swelling was noted over the anteromedial aspect of right arm with gross restriction of range of motion of the shoulder and elbow [Table/Fig-3]. Neck muscles on the posterior aspect were found to be thickened with gross limitation of range of motion. Magnetic resonance imaging (MRI) of cervical spine confirmed the presence of calcified ligamentum nuchae from occiput to seventh cervical vertebra suggestive of Myositis ossificans progressiva [Table/Fig-4].
Clinical picture and dorso-plantar radiograph of both feet showing microdactyly, monophalangeal great toe with hallux valgus deformity, short and stubby first metatarsus with relative overgrowth of the distal epiphyseal ossification center

Antero-posterior radiograph of both thighs and legs showing short and wide necks of both femora with widening of the upper metaphysis of bilateral tibiae giving a false appearance of sessile exostosis / pseudo-exostosis

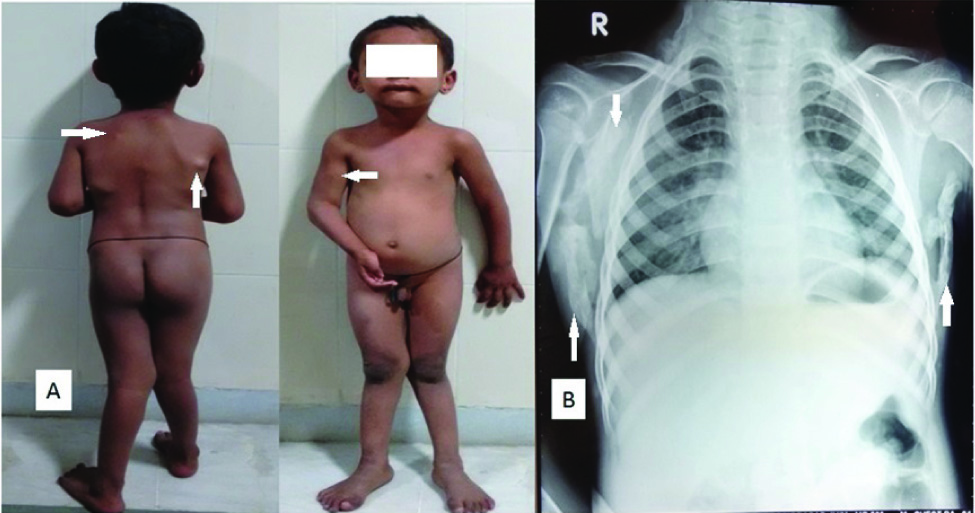
a) Clinical photographs showing multiple nodular swellings on both the scapulae with fixed flexion deformity of right elbow and a nodular mass over the antero-medial aspect of right upper arm. b) Chest radiograph showing rounded to linear ossification along the lateral chest wall surrounding both the scapulae
Sagittal and axial sections of the mri of cervico-dorsal spine showing heterotopic ossification of posterior cervical soft tissues and ligamentum nuchae

FOP was considered as a diagnostic possibility. Blood samples of both the parents and child were collected for sequencing the ACVR1gene. A heterozygous p.R206H mutation, which is the most common mutation reported to be associated with FOP, was detected in the child confirming the diagnosis.
A survey was conducted among 50 orthopaedic surgeons working at various hospitals and institutes regarding the awareness of association of short great toes with congenital hallux valgus and heterotopic ossification with FOP, genetic evaluation for confirmation of the diagnosis and prevention of iatrogenic injury. Six percent of the surveyed consultants were aware of the above features of FOP indicating the necessity for creating awareness about this dreadful rare entity.
Discussion and Literature Review
Fibrodysplasia ossificans progressiva (FOP) also known as Myositis ossificans progressiva is a severely disabling progressive disorder of connective tissues characterized by congenital malformations of the great toes and extensive extra-skeletal heterotopic ossification in explicit anatomic locations [1–4]. It is a rare autosomal dominant genetic disorder with a worldwide prevalence of 1in 2 million population without any racial or geographic preponderance. Nearly 90% of patients with FOP are misdiagnosed, 67% undergo invasive procedures for diagnosis and treatment and more than 50% end up with life long disability [2,5].
FOP occurs due to a spontaneous de novo mutation of the gene encoding activin receptor 1A (ACVR1) - a bone morphogenetic protein (BMP) type1 receptor signalling endochondral ossification. FOP may also be inherited in an autosomal dominant pattern with complete penetrance [2]. Majority of the affected cases reported till date have been found to have a specific R206H (Arginine to Histidine substitution at amino acid position 206) in the gene leading to enhanced BMP signalling [1]. Mutations at other positions of this gene are far less common. This R206H mutation in the ACVR1 gene is regarded as one of the most specific disease causing mutations in the human genome and confirmation of a heterozygous mutation of ACVR1 gene is diagnostic of FOP [6].
Bilateral hallux valgus with microdactyly and monophalangism is classically noted in FOP. In addition to the great toe malformations children with FOP often have less penetrant skeletal aberrations such as short and broad femoral neck, exostosis over proximal tibia, malformed external ear etc. Presence of these lesions singly or in combination supplements the diagnosis of FOP in a child with malformed great toes [2,7,8]. Progressive heterotopic ossification of the connective tissues leads to severe immobility, weight-loss, lung complications and cor-pulmonale from thoracic insufficiency [9]. Majority of FOP cases will be wheelchair bound and dependent by second decade of life and succumb to respiratory complications [1].
Paediatricians and orthopaedic surgeons are often among the first consultants to examine a child with congenital malformation of great toes which is the characteristic feature of FOP [2,6]. Neck stiffness has been an early sign at presentation prior to the appearance of mass lesions [1]. Toe malformations may also be a feature of variety of causes including isolated congenital malformations, synostosis, symphalangism, brachydactyly and juvenile unions. A differential diagnosis of hereditary multiple exostosis, Klippel-Feil syndrome, scleroderma, ankylosing spondylitis is often considered for neck stiffness and multiple swellings [10]. Early diagnosis of FOP is most often missed on evaluation of a child with bilateral congenital great toe malformations and painful rapidly progressive soft tissue swellings [2]. FOP lesions appear and progress rapidly in a span of hours, exceeding the rate of changes seen in most of the aggressive neoplasms [11,12]. There are many instances of open biopsies performed on these swellings which were reported as sarcomas, aggressive fibromatosis or nodular fasciitis [12]. These lesions tend to progress rapidly following invasive surgical interventions. Recurrence in a wider zone on excision of the heterotopic masses has been noticed consistently [1,2,8]. Hence the diagnosis of FOP can be made on the basis of two classical features namely congenital great toe malformations and rapidly progressive soft tissue masses preceding heterotopic ossification in specific anatomic patterns [1]. Genetic analysis for ACVR1gene mutation is confirmatory [2,8]. Delay in the diagnosis and harmful invasive diagnostic and therapeutic interventions often lead to catastrophic life-long disabilities [5].
There is no proven modality of treatment for FOP till date [2,10]. Prevention of progression by avoidance of deep intramuscular injections, injections into jaws for dental procedures, invasive biopsies and excision procedures for heterotopic masses, manipulations for stiff joints and injuries by falls should be considered seriously [10,13]. Acute and painful flare ups can be managed by short term high dose corticosteroids, short term NSAIDs, bisphosphonates [1] and radiotherapy [3]. The futuristic idea of management of FOP relies on targeted gene therapy.
The remarkable feature of this case is the early and unambiguous diagnosis of FOP with pertinent clinical and radiological findings confirmed by the classical R206H mutation in ACVR1 gene. Invasive diagnostic and therapeutic procedures were thereby avoided. Parents were counselled about the disease process and precautions to be taken for prevention of progression.
Conclusion
FOP is a rare musculoskeletal disorder of genetic origin unlikely to be encountered by most of the orthopaedic surgeons leading to misdiagnosis in majority of cases. Inadvertent iatrogenic injury caused by invasive diagnostic procedures done for confirmation of diagnosis in FOP cases end up in rapid progression of the debilitating disease. Congenital hallux valgus with microdactyly and monophalangeal-great toe in a child should be considered as an early diagnostic tool for FOP even before the onset of mass lesions. Confirmation of the diagnosis can be done by genetic analysis of ACVR1 mutation. Unlike many other disorders the principle of primum non nocere i.e. first, do no harm and “masterly inactivity” should be adopted for appropriate management of FOP patients. Prevention of injury by all means, medical management of acute painful flare-ups and rehabilitation should be the goal in the treatment of FOP cases.
Consent
Informed consent of the parents was obtained for publication of case details including clinical photographs.
[1]. Pignolo RJ, Shore EM, Kaplan FS, Fibrodysplasia ossificans progressiva: clinical and genetic aspectsOrphanet J Rare Dis 2011 6:80doi: 10.1186/1750-1172-6-80 [Google Scholar]
[2]. Kaplan FS, Xu M, Glaser DL, Collins F, Early diagnosis of fibrodysplasia ossificans progressivaPediatrics 2008 121(5):e1295-300.doi: 10.1542/peds.2007-1980 [Google Scholar]
[3]. Smith R, Athanasou NA, Vipond SE, Fibrodysplasia (myositis) ossificans progres siva: clinicopathological features and natural historyQJM 1996 89(6):445-46. [Google Scholar]
[4]. Illingworth RS, Myositis ossificans progressiva (Munchmeyer’s disease). Brief review with report of two cases treated with corticosteroids and observed for 16 yearsArch Dis Child 1971 46(247):264-68. [Google Scholar]
[5]. Kitterman JA, Kantanie S, Rocke DM, Kaplan FS, Iatrogenic harm caused by diagnostic errors in fibrodysplasia ossificans progressivaPediatrics 2005 116(5):e654-61.Epub 2005 Oct 17 [Google Scholar]
[6]. Shore EM, Xu M, Feldman GJ, Fenstermacher DA, A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressivaNat Genet 2006 38(5):525-27.Epub 2006 Apr 23 [Google Scholar]
[7]. Kaplan FS, Le Merrer M, Glaser DL, Pignolo RJ, Fibrodysplasia ossificans progressivaBest Pract Res Clin Rheumatol 2008 22(1):191-205.doi: 10.1016/j.berh.2007.11.007 [Google Scholar]
[8]. Raees-Karami SR, Jafarieh H, Ziyayi V, Shekarriz Foumani R, Aghighi Y, Evaluation of 20 years experience of fibrodysplasia ossificans progressiva in Iran: lessons for early diagnosisand preventionClin Rheumatol 2012 31(7):1133-37.doi: 10.1007/s10067-012-1968-6. Epub 2012 Apr 20 [Google Scholar]
[9]. Kaplan FS, Glaser DL, Thoracic insufficiency syndrome in patients with fibrodysplasia ossificans progressivaClin Rev Bone Miner Metab 2005 3:213-16. [Google Scholar]
[10]. Hughes AW, Monsell F, Gargan M, Fibrodysplasia ossificans progressivaCurrent Orthopaedics 2008 22:48-51. [Google Scholar]
[11]. Pignolo RJ, Suda RK, Kaplan FS, The fibrodysplasia ossificans progressiva lesionClin Rev Bone. Miner Metab 2005 3(3– 4):195-200. [Google Scholar]
[12]. Kaplan FS, Tabas J, Gannon FH, Finkel G, Hahn GV, Zasloff MA, The histopathology of fibrodysplasia ossificans progressiva: an endochondral processJ Bone Joint Surg 1993 75(2):220-30.[PubMed: 7678595] [Google Scholar]
[13]. Glaser DL, Kaplan FS, Treatment considerations for the management of fibrodysplasia ossificans progressivaClin Rev Bone Miner Metab 2005 3(3–4):243-50. [Google Scholar]